Publications
The fulltext of publications might not be freely accessible but require subscription. Please request reprints at contact.cube@univie.ac.at.
Publications in peer reviewed journals
3 Publications found
Viruses comprise an extensive pool of mobile genetic elements in eukaryote cell cultures and human clinical samples.
2017 - FASEB J., in press
Abstract:
Viruses shape a diversity of ecosystems by modulating their microbial, eukaryotic, or plant host metabolism. The complexity of virus-host interaction networks is progressively fathomed by novel metagenomic approaches. By using a novel metagenomic method, we explored the virome in mammalian cell cultures and clinical samples to identify an extensive pool of mobile genetic elements in all of these ecosystems. Despite aseptic treatment, cell cultures harbored extensive and diverse phage populations with a high abundance of as yet unknown and uncharacterized viruses (viral dark matter). Unknown phages also predominated in the oropharynx and urine of healthy individuals and patients infected with cytomegalovirus despite demonstration of active cytomegalovirus replication. The novelty of viral sequences correlated primarily with the individual evaluated, whereas relative abundance of encoded protein functions was associated with the ecologic niches probed. Together, these observations demonstrate the extensive presence of viral dark matter in human and artificial ecosystems.-Thannesberger, J., Hellinger, H.-J., Klymiuk, I., Kastner, M.-T., Rieder, F. J. J., Schneider, M., Fister, S., Lion, T., Kosulin, K., Laengle, J., Bergmann, M., Rattei, T., Steininger, C. Viruses comprise an extensive pool of mobile genetic elements in eukaryote cell cultures and human clinical samples.HoloVir: A Workflow for Investigating the Diversity and Function of Viruses in Invertebrate Holobionts.
2016 - Front Microbiol, 822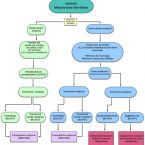
Abstract:
Abundant bioinformatics resources are available for the study of complex microbial metagenomes, however their utility in viral metagenomics is limited. HoloVir is a robust and flexible data analysis pipeline that provides an optimized and validated workflow for taxonomic and functional characterization of viral metagenomes derived from invertebrate holobionts. Simulated viral metagenomes comprising varying levels of viral diversity and abundance were used to determine the optimal assembly and gene prediction strategy, and multiple sequence assembly methods and gene prediction tools were tested in order to optimize our analysis workflow. HoloVir performs pairwise comparisons of single read and predicted gene datasets against the viral RefSeq database to assign taxonomy and additional comparison to phage-specific and cellular markers is undertaken to support the taxonomic assignments and identify potential cellular contamination. Broad functional classification of the predicted genes is provided by assignment of COG microbial functional category classifications using EggNOG and higher resolution functional analysis is achieved by searching for enrichment of specific Swiss-Prot keywords within the viral metagenome. Application of HoloVir to viral metagenomes from the coral Pocillopora damicornis and the sponge Rhopaloeides odorabile demonstrated that HoloVir provides a valuable tool to characterize holobiont viral communities across species, environments, or experiments.Challenges in RNA virus bioinformatics.
2014 - Bioinformatics, 13: 1793-9
Abstract:
Computer-assisted studies of structure, function and evolution of viruses remains a neglected area of research. The attention of bioinformaticians to this interesting and challenging field is far from commensurate with its medical and biotechnological importance. It is telling that out of >200 talks held at ISMB 2013, the largest international bioinformatics conference, only one presentation explicitly dealt with viruses. In contrast to many broad, established and well-organized bioinformatics communities (e.g. structural genomics, ontologies, next-generation sequencing, expression analysis), research groups focusing on viruses can probably be counted on the fingers of two hands.
The purpose of this review is to increase awareness among bioinformatics researchers about the pressing needs and unsolved problems of computational virology. We focus primarily on RNA viruses that pose problems to many standard bioinformatics analyses owing to their compact genome organization, fast mutation rate and low evolutionary conservation. We provide an overview of tools and algorithms for handling viral sequencing data, detecting functionally important RNA structures, classifying viral proteins into families and investigating the origin and evolution of viruses.
Release announcements
News
VOGDB website launched
03.03.17
Latest publications
Contact
- +43 1 4277 76681
- +43 1 4277 876681
- contact.cube@univie.ac.at